RT-PCR實驗操作流程
更新時間:2023-07-17 點擊次數:483次RT-PCR實驗操作流程
上海信帆生物提供 PCR實驗代測,實時熒光定量PCR實驗代測,RT-PCR代測。客戶只需要提供樣本,其他所有試劑都由本公司提供。一般7-10天出結果,提供原始數據,分析數據,實驗報告,實驗室所用儀器型號,圖片,動力擴增曲線等。
RT-PCR實驗外包,mRNA、miRNA、lncRNA、DNA實驗代測
服務內容:
1.制定個性化實驗方案:根據不同的實驗目的確定實驗方案、選定合適的內參;
2.檢測類別:mRNA、miRNA、lncRNA、DNA;
3.樣本抽提及質檢:對客戶提供樣本進行RNA抽提,并利用NanoDrop進行樣本質檢;
4.定量PCR預實驗:包括樣本反轉錄為CDNA、配置PCR反應體系及進行Real-TimePCR反應;
5.正式定量PCR實驗:根據預實驗結果進行正式實驗;
6.數據分析:采用2- △△CT法進行分析,比較不同樣本間差異。
服務要求:
1.請提供組織樣本,細胞樣本,血漿或血清樣本,totalRNA;
2.請提供已知的全長基因序列或GeneBank號;
3.請提供盡可能詳細的背景資料:DNA/RNA來源、基因豐度等。
結果內容:整理好的報告,樣本收到之日起5個工作日內反饋質檢結果。
結果展示:
樣本質檢圖
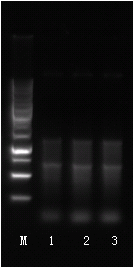
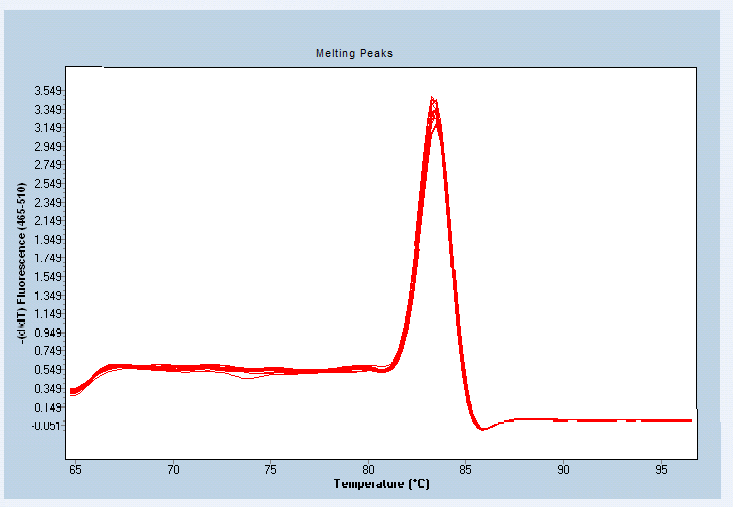
溶解曲線:
擴增曲線

數據統計
每個指標有2張圖。一張是擴增曲線,另一張是熔解曲線,用來證明PCR擴增中無非特異性擴增
送樣運輸要求:
1. 樣品處理
a. 動物組織:常規組織,脂肪組織,結締組織,纖維化組織適當增加。將組織剪切成合適大小后(建議組織塊長寬高均不大于0.5 cm),然后迅速浸泡于5~10倍體積的RNAsolidTM試劑中。(注意:已浸入RNAsolidTM試劑中的組織經平衡一段時間后,可從溶液中取出,切成更小的組織塊,再重新放回RNAsolidTM試劑中;有些比較弱的液體滲透性組織如骨頭等,不適合用RNAsolidTM試劑進行RNA保護。)
b.植物組織:新鮮的葉、果肉、種子最少100 mg。將嫩葉,嫩莖組織切成合適的大小(建議長寬高均不大于0.5 cm)后,然后迅速浸泡于5~10倍體積的RNAsolidTM試劑中。(注意:某些植物組織天生具有的屏障,如葉子表面的蠟層可阻礙RNAsolidTM試劑的滲透,對于此類組織,通常需破壞這些屏障層以允許溶液的浸透。)
c.細胞等樣本:收集不小于10^6個細胞的沉淀(用PBS清洗1-2次)。之后迅速加入5~10倍體積的RNAsolidTM試劑。
d.酵母、細菌等樣本:離心棄上清,收集小米粒大小的單菌體沉淀,然后迅速加入適量的RNAsolidTM試劑浸沒菌體沉淀。
e. RNA樣品: OD260/280為1.8-2.0,濃度≥100 ng/μL,體積≥20μL,干冰運輸。
f.cDNA:cDNA原液≥20 μL,干冰運輸。
Real -Time PCR,即實時監測PCR擴增產物并進行解析的方法,它是在PCR反應體系中加入熒光物質,并通過Real -Time PCR檢測系統對PCR反應進程中的熒光信號強度進行實時監測,終對實驗數據進行分析處理的方法。Real -Time PCR自1996年由美國Applied Biosystems公司推出以來,廣泛應用于生物研究的各個領域。目前主要有SYBR Green 染料、Taqman探針法兩種。
PCR實驗代測,熒光定量PCR代測服主要儀器及耗材
旋渦振蕩器 :上海青浦瀘西儀器廠產品
手握式電動勻漿機:德國FLUKO 公司產品
低溫冷凍離心機:Sigma(3K15)公司產品
Real-time檢測儀:ABI (ABI-7300)公司產品
超純水分離系統:美國LABCONCO公司
離心管及10μL、200μL、1000μL槍頭等常規耗材均為國產;RNA提取過程中所需的離心管、槍頭等耗材經過0.1%的DEPC水浸泡過夜,121℃滅菌30min,烘干后備用。
PCR實驗代測,熒光定量PCR代測服實驗室試劑的操作
1)所用的所有溶液都應該沒有核酸和(或)核酸酶(DNase和RNase)污染。
2)所有PCR試劑中使用的水都應該是高質量的-新鮮蒸餾的去離子水,用0.22μm過濾的,并且是高壓滅菌。
3)在20℃到25℃貯存的試劑建議加點像疊氮NA一類的抗微生物劑,在擴增試劑或樣品制備試劑中加入0.025%的疊氮NA不抑制擴增反應。
4)所用試劑都應該以大體積配制,實驗一下看試劑是否滿意,然后分裝成僅夠一次使用的量進行貯存。
5)所有試劑和樣品準備過程中都要使用一次性滅菌的瓶子和管子。
6)新配制的試劑在用于準備新的標本之前應該加以檢驗。
7)樣品準備和前PCR區所使用的移液管在不使用時都應該小心保存。
本程序適用于自動PCR操作,可適用于大多數情況,所用酶是不含核酸外切酶活性的熱穩定聚合酶,如Taq聚合酶等。
引物(各50pmol) 0.2mmol/L dNTPs(必須使用高質量的dNTPs,這一點非常重要。dNTPs經反復化凍后會發生降解,因此應分成小份保存。應注意混合物中四種dNTP的量要相等) 模板DNA:約0.05~1mg基因組DNA或0.002~0.02mg質粒DNA Taq DNA聚合酶(Perkin Elmer, Norwalk, Connecticut) 10×PCR緩沖液(500mmol/L KCl,15mmol /L MgCl2,100mmol/L Tris·HCl,pH 8.3) 礦物油
電泳所需試劑
【儀器設備】
PCR擴增儀
電泳裝置
微量離心機
微量移液器(1~20ml和20~200ml)
0.5ml Eppendorf 管
紫外線觀察裝置及照相設備
操作程序
1.在無菌的Eppendorf管內加入以下反應物:
2.93℃ 反應2 min后開始以下循環:
93℃變性反應1 min;
50℃退火反應1 min;
72℃延伸反應3 min。
經過17~35循環后,后一個循環72℃增加5 min。循環結束后反應產物置于40℃保存。
3.取1~5ml反應產物走凝膠電泳,經溴化乙錠染色檢測擴增的情況。
應注意的問題及其解決方法
1.PCR反應條件的優化
要優化特定的PCR反應,有必要試用不同的反應組分和循環參數。表2-1列出了添加或改變某一試劑濃度對反應結果的影響,從中大家可以得到一些線索以改進反應條件,提高PCR產量和/或特異性,一般情況下,只須改變一兩個參數就行了,如pH值和MgCl2濃度。表2-2給出了循環參數對PCR結果的影響。
表2-1 PCR各反應組分濃度對產物特異性和產量的影響
| 反應組分 | 提高產量 | 提高特異性 |
| 模板濃度 | 增加 | 減少 |
| 引物濃度 | 高至75 pmol | 低至15 pmol |
| 引物長度 | - | 增加 |
| dNTPs | 各1 mmol/L | 各20 mmol/L |
| Tris-HCl (10 mmol/L pH8) | pH高至10* | pH高至10* |
| MgCl2 | 高至4 mmol/L | 1.5 mmol/L |
| DMSO | - | 10%** |
| 甘油 | - | 2% |
| 甲酰胺 | - | 5% |
| Taq DNA聚合酶*** | 高至5U | 0.5U |
* 效果可能不可預測
** 添加10% DMSO時,Taq DNA聚合酶的活性會被抑制47%,因此反應加大Taq DNA聚合酶的用量(即5U)予以補償
*** Perkin Elmer, Norwalk, Connecticut
表2-2 可改變以提高PCR產物的特異性和/或產量的循環參數
| 反應條件 | 提高產量 | 提高特異性 |
| 循環數 | 多35個 | 減至25個 |
| 變性* | 增至1 min | - |
| 引物退火** | 降至45℃ | 升至72℃ |
| 引物延伸*** | 在后期循環中延長 | 維持30s |
* 使用基因組DNA作模板時,開始幾個循環變性應于97℃進行
** 假定Tm值為60℃
*** 延伸時間依產物大小而定:1000bp的產物延伸30s即可,產物更長時,按每1000 bp延伸0.5 min計,在后期循環中增加延伸時間可以提高擴增效率
2.引物的設計
毫無疑問,引物的堿基組成,長度及其靶序列的同源性是決定PCR成敗的首要因素,選擇設計引物在一定程度上仍然要靠經驗,對于某一對引物而言尚無通用的規則可嚴,但應遵守以下原則:
(1)一般性原則:
①長度:15~30bp,其有效長度 [Ln=2 (G+C)+(A+T)] 一般不大于38,否則PCR的適延伸溫度會超過Taq酶的佳作用溫度(74℃),從而降低產物的特異性。
②G+C含量:應在45%~55%之間,PCR擴增中的復性溫度一般是較低Tm值引物的Tm值減去5~10度。引物長度小于20時,其Tm值等于4×(G+C)+2×(A+T)。
③ 堿基分布的隨機性:應避免連續出現4個以上的單一堿基。尤其是不應在其3’端出現3個的連續的G或C,否則會使引物在G+C富集序列區錯誤引發。
④ 引物自身:不能含有自身互補序列,否則會形成發夾樣二級結構。
⑤引物之間:兩個引物之間不應有多于4個的互補或同源堿基,不然會形成引物二聚體,尤應避免3’端的互補重疊。
⑥特異性:與非特異擴增序列的同源性應小于70%,或少于連續8個的互補堿基。
(2)引物的3’端:
引物的3’端很大程度上影響Taq酶的延伸效應,除特殊情況(AS-PCR)外,3’端不應發生錯配。S. Kwok等發現,引物的3’端發生錯配時,A:A使產量下降至1/20,A:G或C:C下降至1/100。當末位堿基是T時,即使錯配也能引發鏈的合成,而為A時錯配的引發效率低,G、C居間。
因此,引物的3’端堿基好選用A、G、C,而盡可能地避免選用T,尤應避免連續出現2個以上的T。
此外,如果擴增編碼區域,引物的3’端不要終止于密碼子的第3位,因此處易發生簡并,從而影響擴增特異性。
(3)避開引物的二級結構區:
某些引物無效的原因是引物重復區二級結構的影響,因此選擇擴增片段時應注意避開二級結構區,有些計算機軟件可幫助確定該區域,如不能避開,則可用7- deaza-2’-脫氧GTP取代dGTP,有助于PCR成功。
目前在引物選擇中已廣泛應用計算機軟件,從EMBL或Genebank中查找有關基因序列,并依據上述原則對所選用引物進行評價。
3.出現異常結果時的解決思路
在做PCR反應的過程中,由于其酶促反應的本質,影響終結果的因素較多,所以出現一些非預料的結果是可以理解的。下面簡單地總結一下在PCR反應結果出現異常時我們應該采取的措施。
(1)電泳檢查沒有 PCR產物
a.需檢查一下Taq DNA聚合酶是否失活,或是活性太低,還需查一下在加樣過程中是否向體系中加過Taq DNA聚合酶;
b.需檢查一下所使用的引物,看其序列設計是否正確,是否堿基組成不平衡;
c.需要檢查一下PCR反應過程中模板是否變性充分,尤其在前幾個循環中是否變性充分;可以適當地提高模板變性的溫度或是適當延長變性的時間;
d.需檢查一下在PCR反應體系中是否有Taq DNA聚合酶的抑制劑,如SDS污染等等;
e.需檢查一下模板中是否有蛋白酶和核酸酶的污染,可以預先加熱使之失活。
(2)電泳檢查出現引物二聚體區帶
a.需檢查一下兩個引物的3’端是否互補,或者是否有相類似的回文結構;
b.需檢查一下使用的引物是否太短,可以予以適當的延長;
c.需檢查模板的用量,模板以10-4個拷貝為佳,模板太少會造成引物相對過量;
d.適當地降低引物的濃度,引物濃度過高是出現引物二聚體的主要原因;
e.循環次數太多也易形成引物二聚體,可以適當地減少循環數;
f.可以將復性溫度提高一些以減少引物二聚體形成。
(3)電泳檢測PCR產物呈片狀(smear)
a.需檢查一下Taq DNA聚合酶的用量,適當地減少Taq酶的量有助于特異性條帶擴增;
b.可以提高反應的退火溫度,或者直接采用兩個溫度的PCR(68℃退火和延伸,94℃模板變性);
c.適當地降低Mg 2+ 的濃度也可起到降低非特異性擴增可能性的作用;
d.適當地減少反應退火的時間和延伸的時間;
e.適當地減少循環次數也會有效果;
f.可以嘗試使用巢式引物PCR(nested primer PCR)。
(4)電泳檢測PCR產物出現其它非特異性條帶
a.需檢查一下引物的3’端是否有連續排列的G或C;
b.可以適當地提高退火溫度或直接采用兩步溫度PCR方法進行擴增;
c.可以降低Taq DNA聚合酶的用量,同時也降低引物的濃度;
d.可以適當地減少退火所用的時間以及延伸反應的時間;
e.可以適當地增加或減少一些Mg 2+ 濃度。
4.假陽性
實驗中設立的陰性對照可提示有無假陽性結果出現。如果一次實驗中的幾個陰性對照中出現一個或幾個陽性結果,提示本次實驗中其它標本的檢測結果可能有假陽性。造成假陽性的原因包括樣品污染、擴增試劑污染、擴增產物交叉污染等。常見的污染來源包括實驗室環境、加樣器、操作中形成的噴霧、DNA抽提儀器、試劑及任何與擴增產物接觸的東西。
預防各種污染的措施主要有:(1)工作區隔離。(2)改進實驗操作,如在加樣過程中避免試劑飛濺、吸頭離心管使用前高壓處理、試劑分裝成小份一次使用后棄去等。(3)操作程序合理化,如后加陽性對照等。
PCR實驗代測,PCR實驗設計,PCR實驗--上海信帆生物代做免疫組化實驗,WB實驗,放免實驗,ELISA實驗等,提供實驗數據,圖片,實驗報告。
RT-PCR實驗操作流程
